Research
There is an abundance of publicly available data about various biological systems, but it can be difficult to draw insight from individual datasets. Our lab develops algorithms that integrate these data to help model and understand complex biological systems. Doing this allows us to investigate many different biological conditions, including those with limited data, such as rare diseases. We recognize that our lab won’t have all the answers, or even all of the questions, so we aim to develop tools and processes that any biologist can reuse. Our approach to research prioritizes transparency, rigor, and reproducibility.
The citations on this page were generated automatically from just identifiers using the Manubot cite utility developed right here in the Greene Lab!
2024

2023






















2022






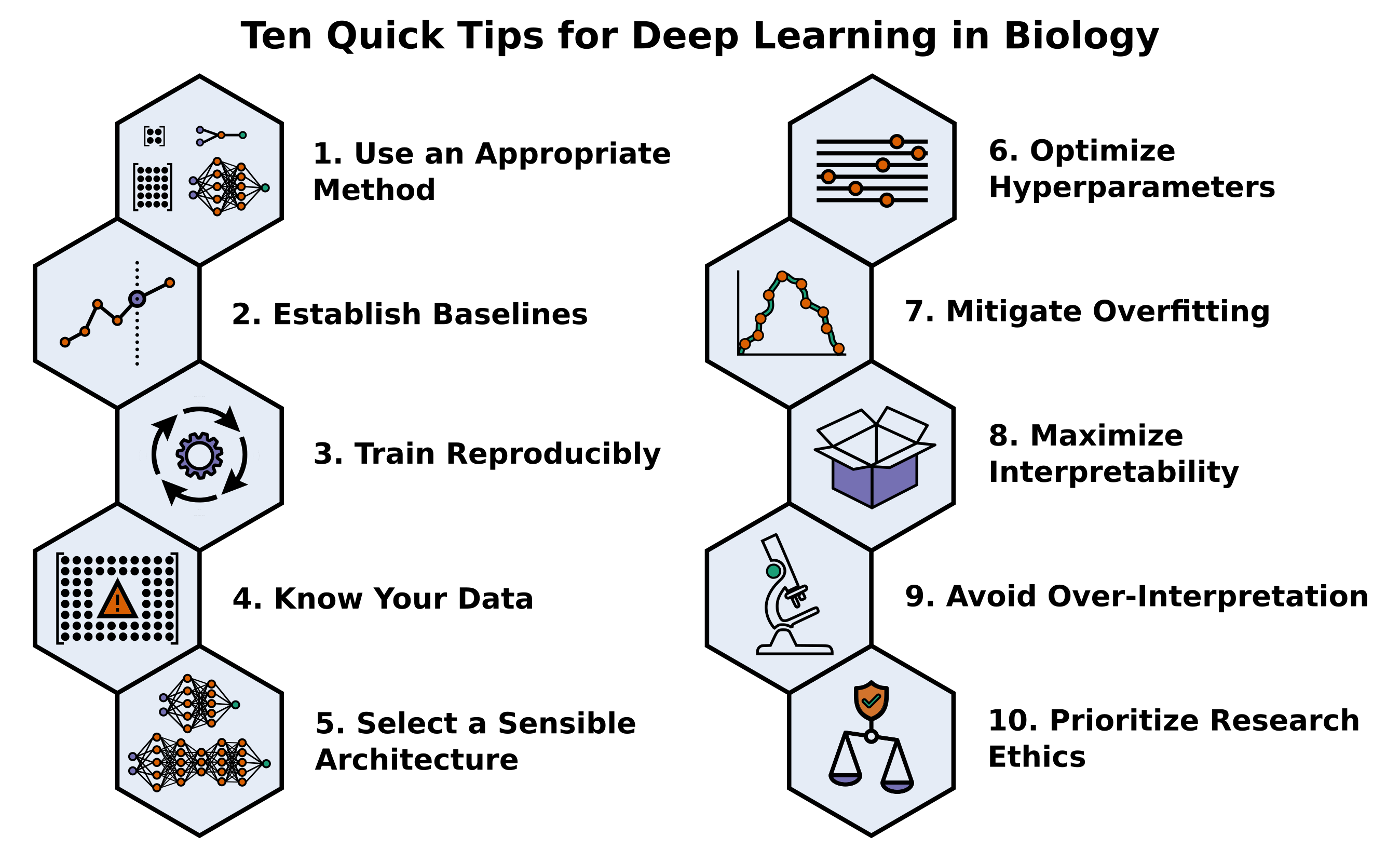




2021






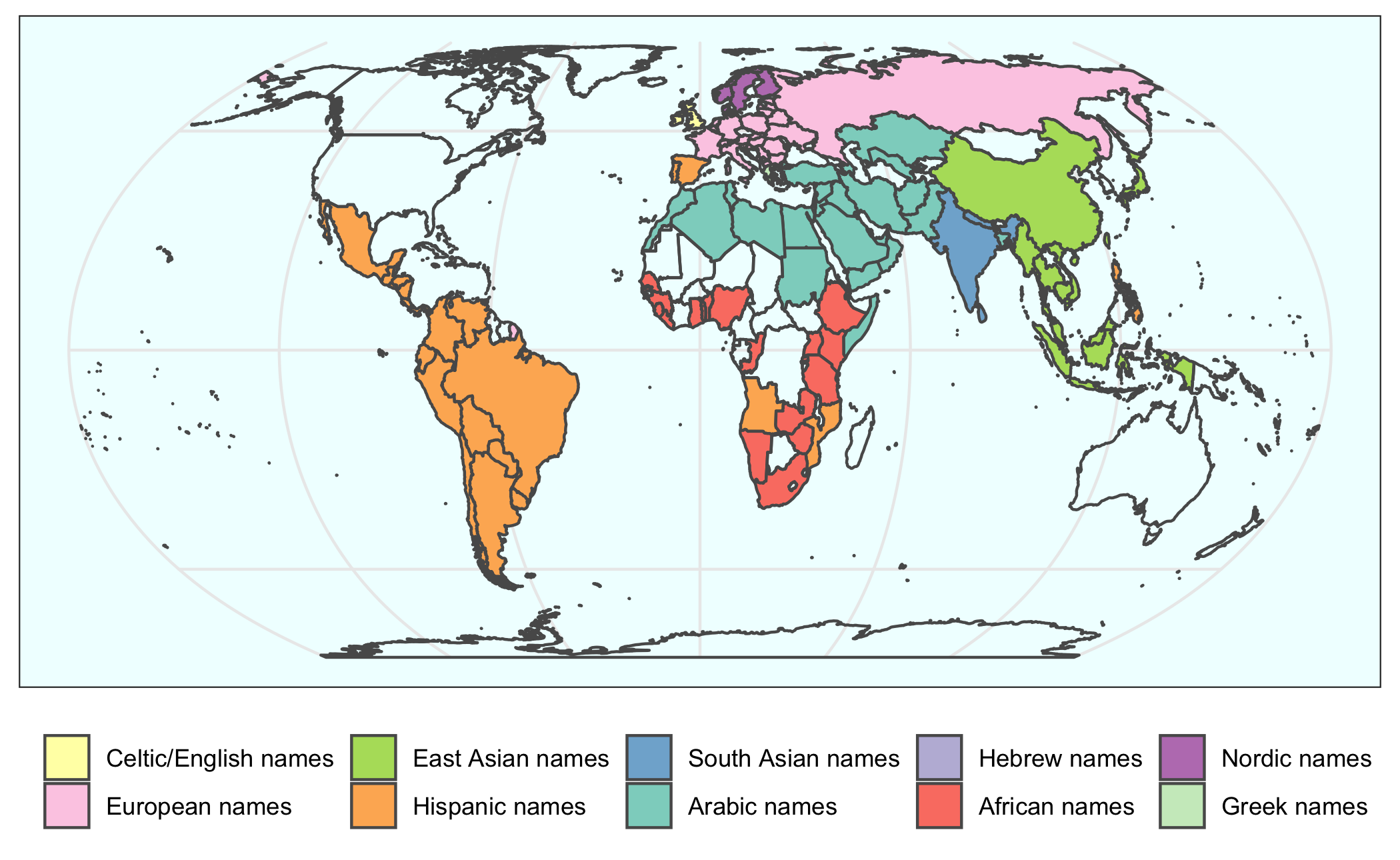


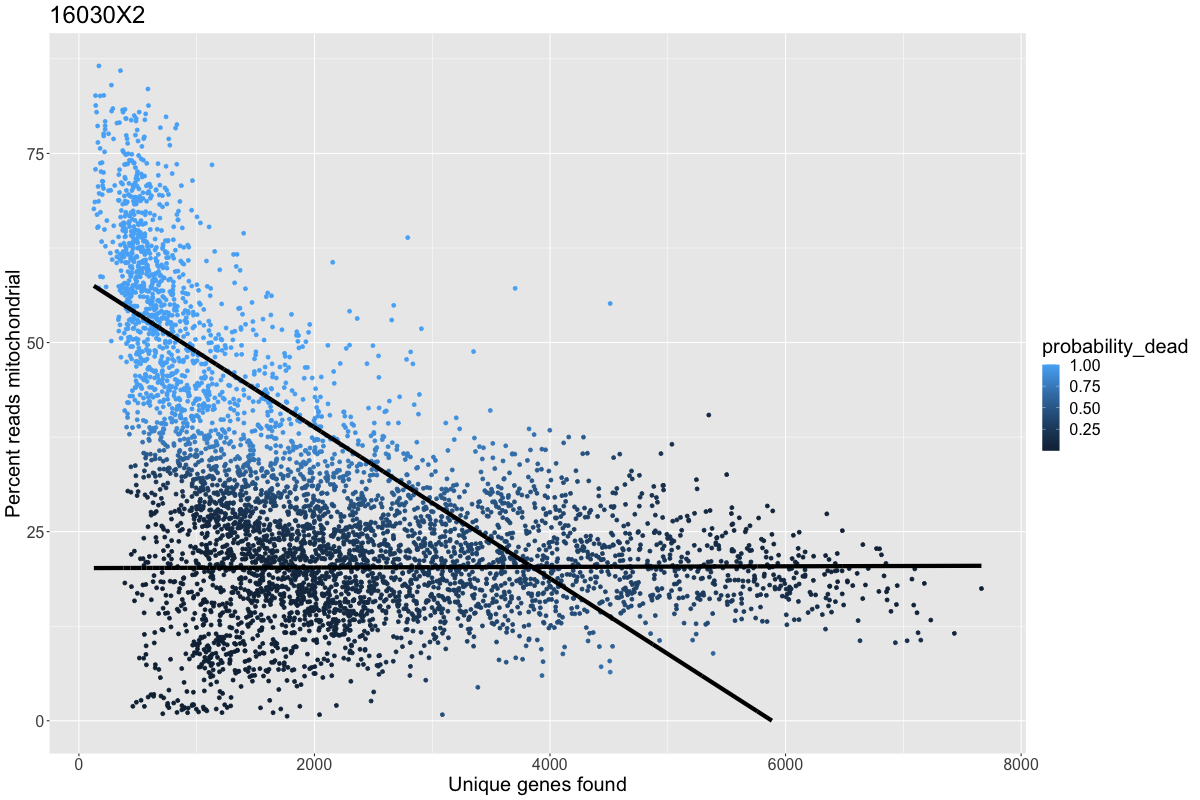
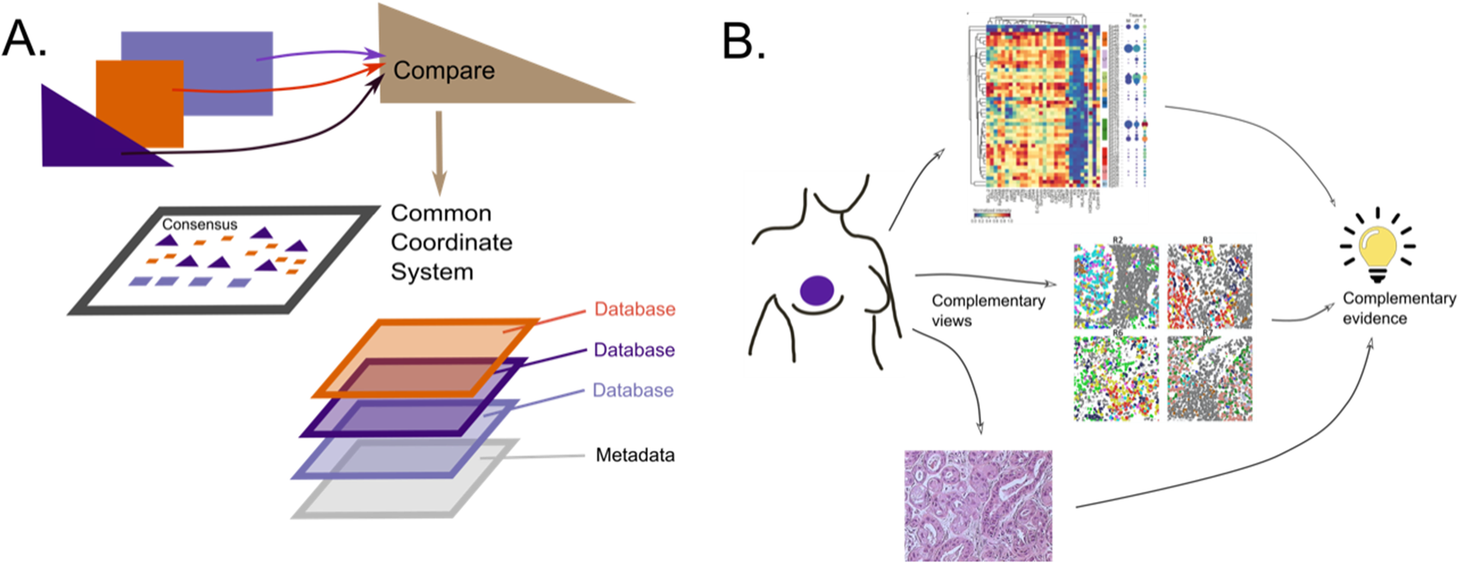







2020







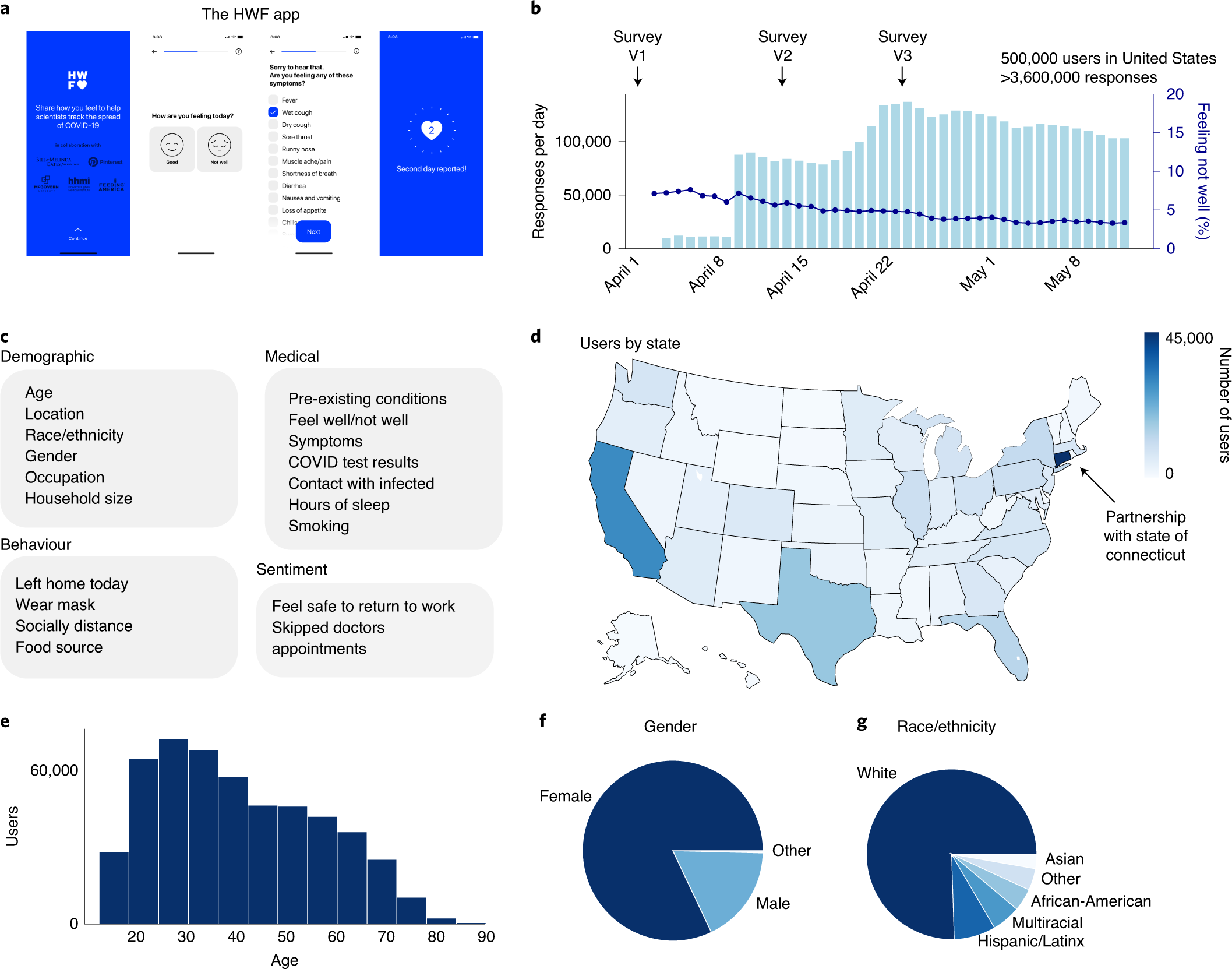



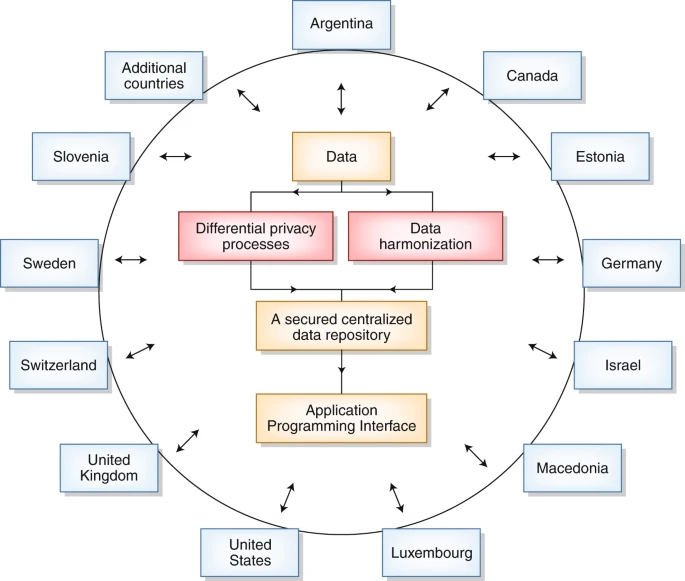


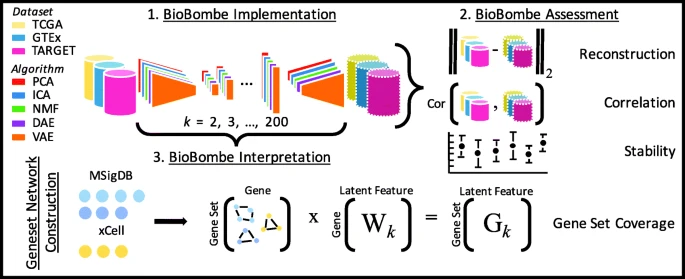

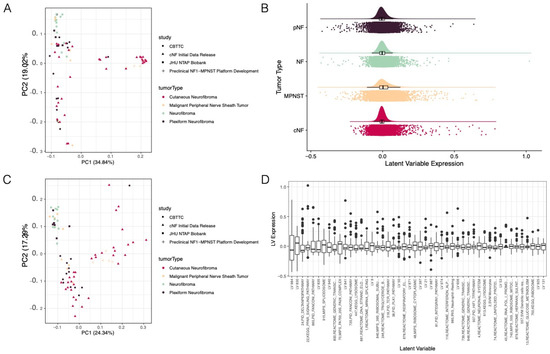


2019
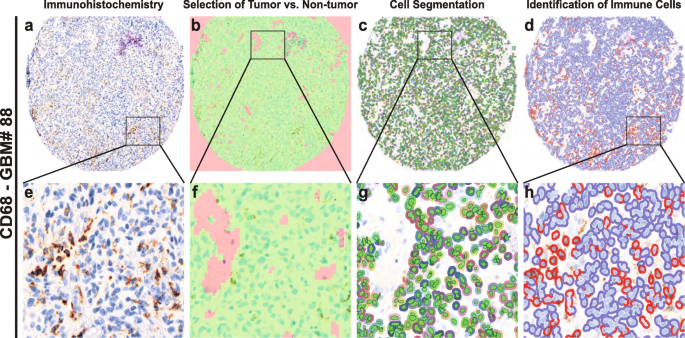
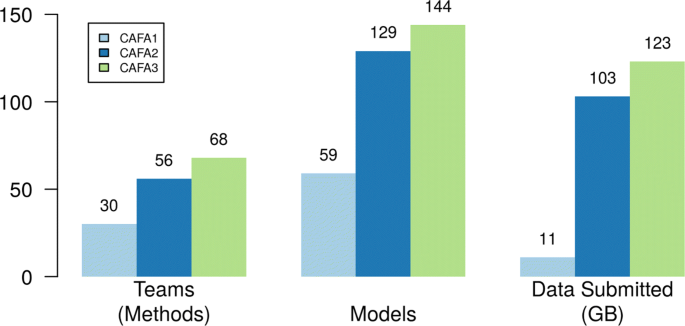








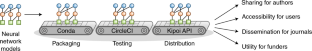




2018
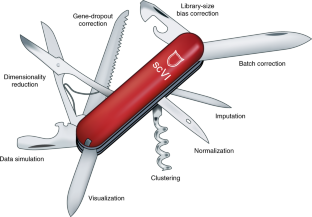




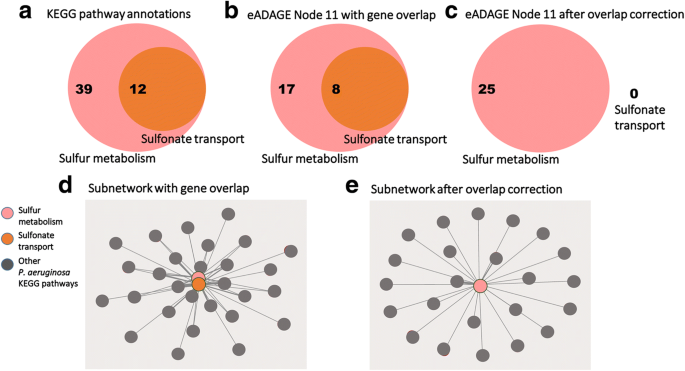







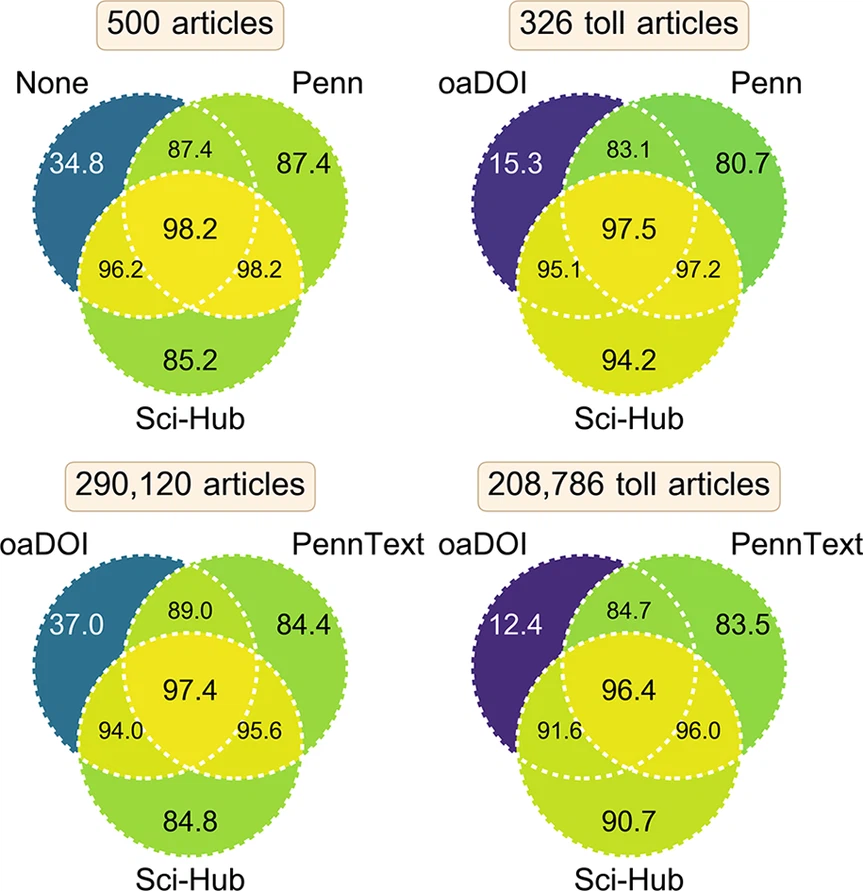


2017
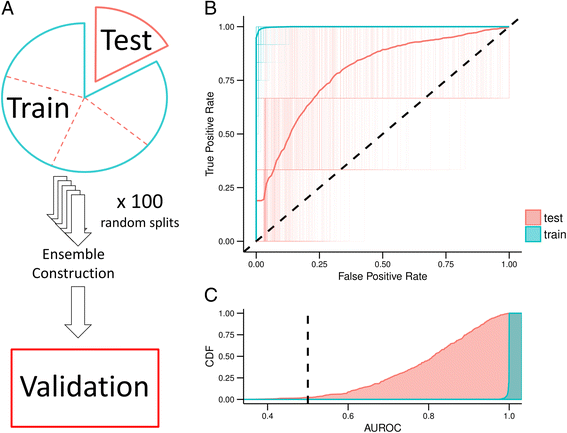
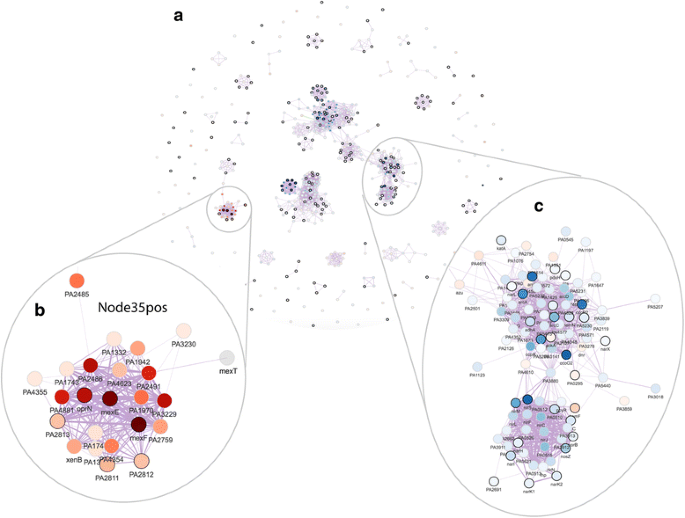


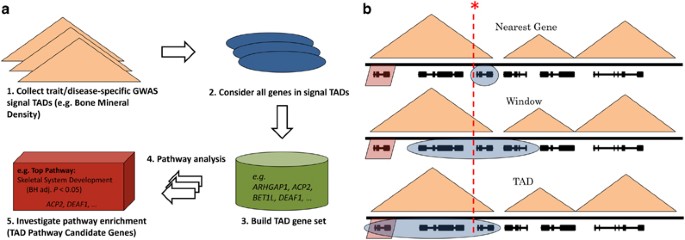

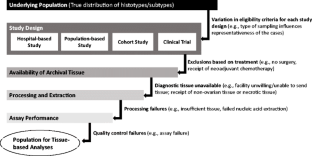


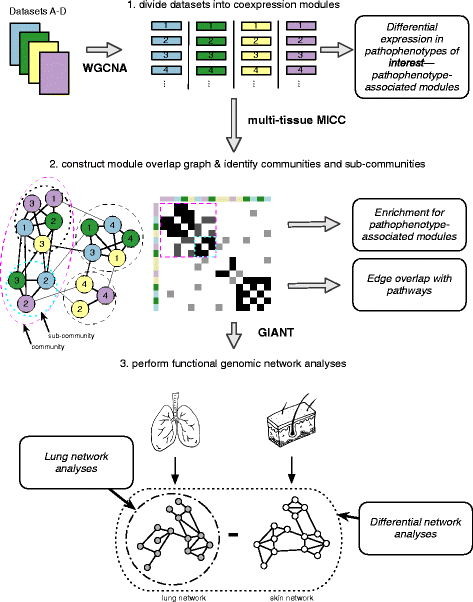
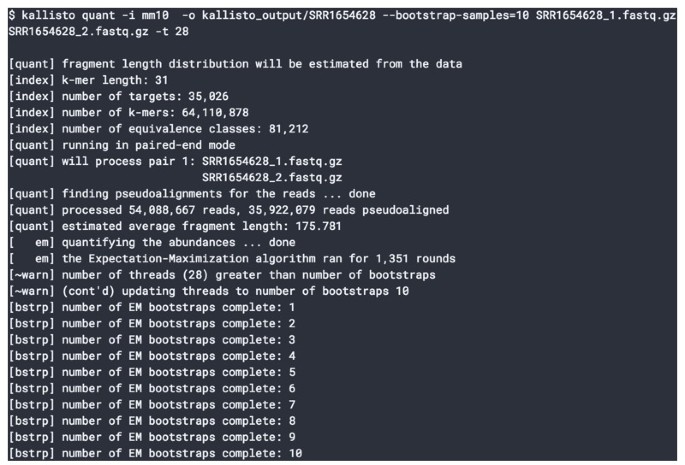

2016













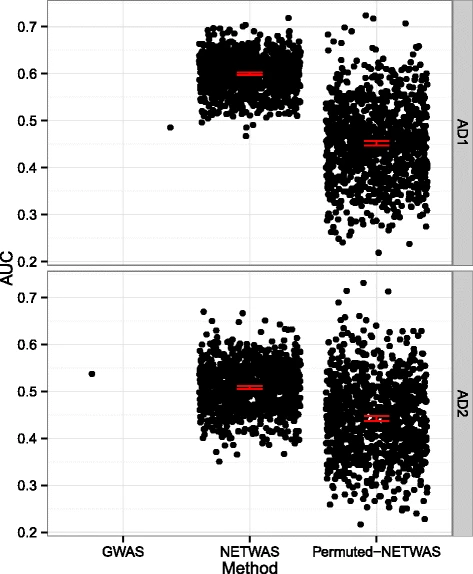

2015
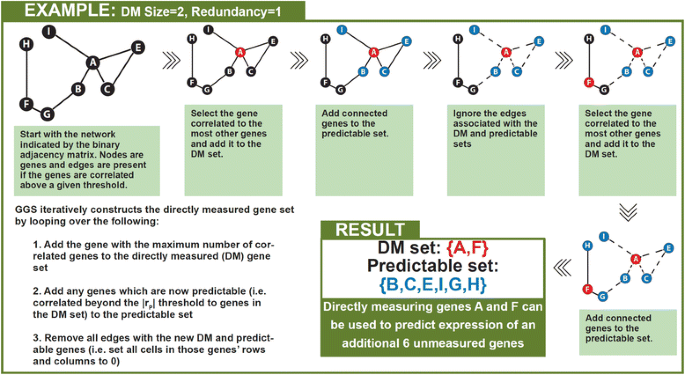



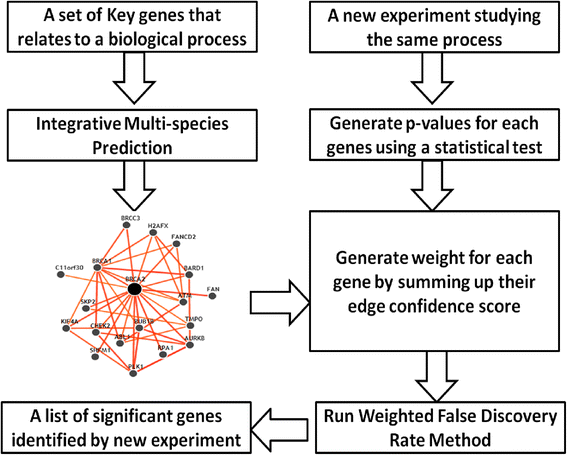
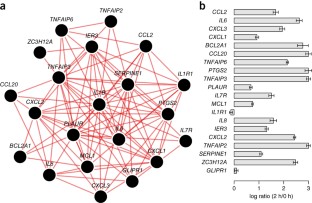



2014

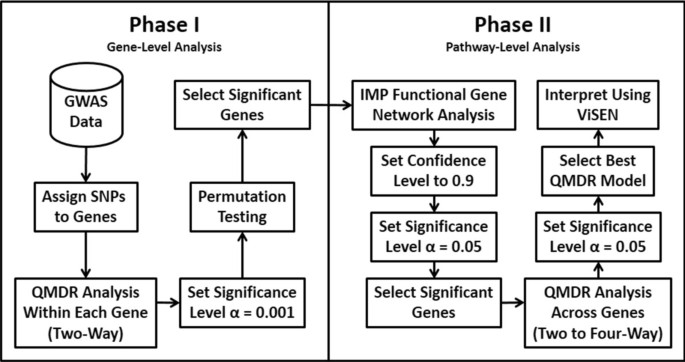
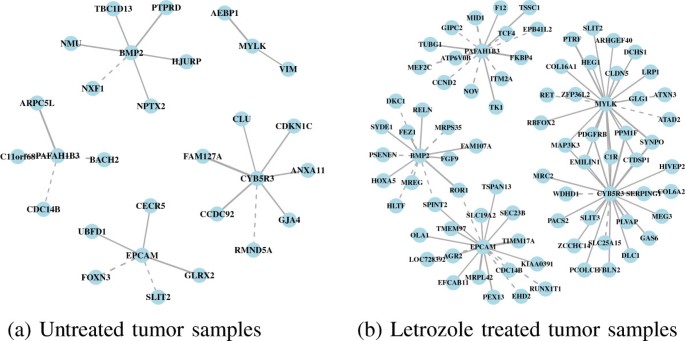
2013


2012

